T. Böhler*, H.J. Girschick**, G. Horneff***
* Medizinischer Dienst der Krankenversicherung Baden-Württemberg
** Univ.-Kinderklinik Würzburg
*** Univ.-Kinderklinik Halle
Neutropenie ist der Mangel an peripher zirkulierenden neutrophilen Granulozyten. Bei der milden Neutropenie liegt die absolute Neutrophilenzahl zwischen 1000 und 1500/μl, bei der mittelschweren zwischen 500 und 1000/μl und bei der schweren unter 500/μl. Zur Berechnung der absoluten Zahl neutrophiler Granulozyten wird die Leukozytenzahl mit dem Ergebnis des Differentialblutbildes (Prozentanteil der segmentkernigen und stabkernigen neutrophilen Granulozyten und deren unreifer Vorstufen) im peripheren Blut multipliziert.
Die Differenzierung von der hämatopoetischen Stammzelle bis zum reifen neutrophilen Granulozyten und deren Auswanderung aus dem Knochenmark in das periphere Blut erfordert die koordinierte Stimulation durch verschiedene Zytokine und Chemokine: Stromal-derived Factor (SDF)-1, Stammzellfaktor (SCF), Granulozyten-Makrophagen-Kolonie-stimulierender Faktor (GM-CSF) und Granulozyten-Kolonie-stimulierender Faktor (G-CSF), Interkleukin-3 (IL-3), IL-6 und IL-8.
Aus Störungen dieser zellulären Entwicklungsprozesse durch angeborene oder erworbene Erkrankungen bzw. Umwelteinflüsse resultieren vielfältige Differentialdiagnosen (Tab. 1). Bei der aplastischen Anämie, dem myelodysplastischen Syndrom, der retikulären Dysgenesie und einer Reihe von primären Immundefekten sowie bei Leukämien, Lymphomen und das Knochenmark infiltrierenden Malignomen ist die Neutropenie Symptom einer Grunderkrankung. Häufig sind in diesen Fällen weitere Zelllinien der Hämatopoese betroffen (Anämie und/oder Thrombozytopenie).
Die isolierte schwere angeborene Neutropenie (Synonym: infantile Agranulozytose) wird bei Neugeborenen bzw. im frühen Säuglingsalter diagnostiziert. Sie ist gekennzeichnet durch eine stark erniedrigte Absolutzahl neutrophiler Granulozyten im peripheren Blut (stets unter 200/μl). Dabei zeigt die Knochenmarkaspiration den Befund eines Ausreifungsstopps der neutrophilen Reihe auf der Stufe der Promyelozyten oder der frühen Myelozyten. Die Zahl der Monozyten und eosinophilen Granulozyten ist vermehrt, die Immunglobulinspiegel der betroffenen Patienten sind auf Grund rezidivierender Infektionen stark erhöht.
Die zyklische (periodische) Neutropenie ist durch eine rekurrierende Störung der Ausreifung hämatopoetischer Stammzellen gekennzeichnet und führt im Abstand von 2 bis 6 Wochen zu wiederkehrenden Phasen der peripheren Neutropenie mit einer Dauer von weniger als 7 Tagen (meist 4-5 Tage). Im Knochenmark findet sich je nach Entwicklungsphase des Zyklus ein unterschiedliches Bild; neben der Granulopoese kann jedoch auch die Thrombopoese und die Hämatopoese gestört sein.
Als Ursachen der schweren angeborenen Neutropenie (OMIM #202700) wurden heterozygote, dominant-negative Mutationen im Gen der Neutrophilen-Elastase ELA2 (OMIM 130130) bzw. des nukleären Zink-Finger-Proteins GFI1 (Growth factor-independent 1; OMIM 600871) identifiziert (Tab. 2). Sie führen zu sporadischen oder autosomal-dominant vererbten Erkrankungsformen (Ancliff et al. 2001, Person et al. 2003). Die Mutationen im GFI1-Gen stören die Repressoraktivität dieses Transkriptionsfaktors und erhöhen damit die Expression von ELA2 in neutrophilen Granulozyten betroffener Patienten (Person et al. 2003). Die Mutationen im ELA2-Gen führen zu einer konstitutiv erhöhten Aktivität der Neutrophilen-Elastase.
Das ELA2-Gen ist nicht nur bei Patienten mit autosomal-dominanter schwerer angeborener Neutropenie mutiert, sondern auch bei Patienten mit milder verlaufender zyklischer Neutropenie (OMIM #162800). Der genaue Pathogenesemechanismus, durch den die erhöhte Enzymaktivität von ELA2 zu einem Mangel reifer neutrophiler Granulozyten im peripheren Blut führt, ist nicht bekannt. Eine direkte Rolle der Neutrophilen-Elastase in der Granulopoese konnte bislang nicht nachgewiesen werden.
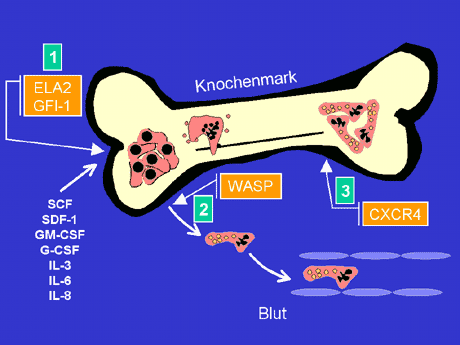
Ein Erklärungsmodell geht von der Beobachtung aus, dass Promyelozyten und Myelozyten mit mutierter oder vermehrt exprimierter ELA-2 eine selektiv gesteigerte Apoptoserate aufweisen. Diese Steigerung des programmierten Zelltodes soll den im Knochenmark beobachteten "Reifungsstopp" erklären (Abb. 1).
Abb. 1: Genetische Störungen der Granulopoese als Ursache für Formen der schweren angeborenen Neutropenie bzw. der zyklischen Neutropenie. Proliferation und Apoptose hämatopoetischer Stamm- und Vorläuferzellen werden ebenso wie die Auswanderung reifer neutrophiler Granulozyten aus dem Knochenmark in das Blut durch Wachstums- und Chemotaxisfaktoren (Zytokine und Chemokine) geregelt. Bei der angeborenen schweren Neutropenie findet sich im Knochenmark eine verstärkte Apoptose von (Pro-) Myelozyten, die als Ausreifungsstopp imponiert. Ursache sind Mutationen im ELA2- bzw. GFI-1-Gen (1), die zu einer erhöhten Aktivität der Neutrophilen-Elastase führen sowie eine konstitutiv-aktivierende Mutation im WASP-Gen (2). ELA2-Mutationen können auch zu einer zyklischen Neutropenie führen, wenn eine ausreichende Regenerationskapazität der Granulopoese besteht. Bei Mutationen im Chemokin-Rezeptor-Gen CXCR4 (3) kommt es zu einer Anhäufung reifer, übersegmentierter neutrophiler Granulozyten im Knochenmark (Myelokathexis). (Abkürzungen: SCF = stem cell factor, SDF-1 = stromal cell-derived factor-1, CSF = colony stimulating factor, GM = granulocyte-macrophage, G = granulocyte, IL = interleukin). (Film)
Steht im Knochenmark eine ausreichende Reserve für die Neubildung von Vorläuferzellen der Granulopoese zur Verfügung, sollen die Mutationen im ELA2-Gen zur zyklischen Neutropenie führen, fehlt diese Regenerationskapazität, resultiert eine schwere chronische Neutropenie. Da identische ELA2-Mutationen einen unterschiedlichen Phänotyp hervorrufen können, wird eine bestimmende Rolle weiterer, bislang nicht bekannter Gene für die Krankheitsausprägung angenommen.
Zwei weitere Formen der schweren chronischen Neutropenie sind bekannt:
· die X-chromosomal rezessiv vererbte Form (X-gebundene schwere angeborene Neutropenie; OMIM #300299) wird durch spezifische, konstitutiv-aktivierende Mutationen im Wiskott-Aldrich-Syndrom-Protein (WASP; OMIM 300392.0012) hervorgerufen;
· die autosomal-rezessiv vererbte infantile Agranulozytose (nach dem Erstbeschreiber der Erkrankung als Morbus Kostmann bezeichnet; OMIM #202700) beruht möglicherweise auf genetisch bedingten Störungen in der Funktion eines zellulären Inhibitors der Neutrophilen-Elastase ELA2; die genaue molekulare Ursache der Erkrankung ist jedoch bislang nicht bekannt.
Das autosomal-dominant vererbte WHIM-Syndrom (Warzen, Hypogammaglobulinämie, Infektionen und Myelokathexis; OMIM #193670) ist gekennzeichnet durch eine periphere Neutropenie mit hyperzellulärem Knochenmark und übersegmentierten, vakuolisierten neutrophilen Granulozyten (sog. Myelokathexis) und eine Erniedrigung der Immunglobulinspiegel (Hypogammaglobulinämie). Es kommt zu gehäuften Infektionen insbesondere im Genitalbereich durch Befall mit Papillomviren (Warzen, Condylomata acuminata, Dysplasie der Zervixschleimhaut). Verursacht wird diese Krankheit durch Mutationen im Gen des Chemokinrezeptors CXCR4 (OMIM 162643). Der Ligand dieses Rezeptors ist das für die Granulopoese wichtige Chemokin SDF-1 (Stromal cell-derived Factor-1).
Bei Nachweis einer Neutropenie muß eine genaue Medikamentenanamnese erhoben werden, um Arzneimittel-induzierte Neutropenien, z.B. nach Antibiotikagabe, ausschließen zu können. Liegt eine milde isolierte Neutropenie ohne sonstige klinische Symptomatik vor, sind kurzfristige Verlaufskontrollen des Blutbilds mit Differentialblutbild und klinische Untersuchung erforderlich. Oft kommt es zur spontanen Remission im Verlauf von 5-6 Wochen. Finden sich wiederholt erniedrigte Neutrophilenzahlen, so liegt eine chronische Neutropenie vor.
Durch zwei Blutbildkontrollen pro Woche über 6 Wochen kann eine zyklische Neutropenie diagnostiziert werden. Zyklische Neutropenien treten alle 2-3 Wochen für die Dauer von ca. 1 Woche auf und werden durch regelmäßige Kontrollen der Neutrophilenzahl über 2 Monate (anfangs 2 x pro Woche, dann 1 x wöchentlich) festgestellt. Entwickeln sich während dieser Zeit Symptome einer zugrundeliegenden Erkrankung oder einer schweren Infektion bzw. fehlt der Anstieg von Neutrophilen im Rahmen eines Infekten, so ist eine weiterführende Diagnostik zum Ausschluß von Immundefekten, malignen Erkrankungen, einer HIV-Infektion oder eines systemischen Lupus erythematodes erforderlich.
Der Nachweis von Autoantikörpern gegen neutrophile Granulozyten spricht für das Vorliegen einer Autoimmunneutropenie. Bestehen neben der Neutropenie gleichzeitig eine Anämie und/oder Thrombopenie oder finden sich Blasten im peripheren Blutausstrich, ist unverzüglich eine Knochenmarkaspiration zum Ausschluß einer malignen Infiltration des Knochenmarks erforderlich. Auch bei Vorliegen einer persistierenden Neutropenie (absolute Granulozytenzahl im peripheren Blut bei Kindern jenseits des ersten Lebensjahres < 1500/μl, bei Säuglingen < 1000/μl über mehr als 4 Wochen) muß eine Knochenmarkaspiration zum Ausschluß einer hämatologischen Systemerkrankung erfolgen. Bei peripherer Neutropenie im Rahmen des WHIM-Syndroms findet sich ein hyperzelluläres Knochenmark durch Retention der reifen neutrophilen Granulozyten.
Eine chronisch-benigne Form der Neutropenie kann angenommen werden, wenn andere Ursachen soweit möglich und der Schwere der Symptomatik entsprechend erforderlich ausgeschlossen sind (Ausschlußdiagnose). Die Glykogenose Typ Ib und das Shwachman-Diamond-Syndrom können anhand richtungsweisender zusätzlicher klinischer und immunologischer Befunde diagnostiziert werden.
Patienten mit chronischen und rezidivierenden Infektionen der Lymphknoten, Haut, Leber, Lunge und des Gastrointestinaltraktes sowohl mit extrazellulären (bekapselte Bakterien) als auch intrazellulären Erregern (z.B. Pilze) weisen gehäuft Störungen der Neutrophilenzahl auf. Die resultierende Immundefizienz betrifft insbesondere die Abwehr gegen gramnegative Bakterien und Staphylokokken. Dementsprechend eingeschränkt ist die lokale Entzündungsreaktion, insbesondere die Eiterbildung. Abszesse enthalten wenig Eiter, Ulzerationen zeigen kaum einen schmierigen Belag.
Die Autoimmun-Neutropenie ist kein primärer Immundefekt. Sie ist jedoch die häufigste Ursache einer persistierenden Neutropenie des Säuglings und Kleinkindes und geht mit gehäuften bakteriellen Infektionen (Otitis) einher, wobei schwere Infektionen selten sind. Bei bakteriellen Infektionen können die Neutrophilenzahlen in den Normalbereich ansteigen. Es muß eine konsequente antibakterielle Therapie, ggf. eine antibakterielle Prophylaxe (Cotrimoxazol) erfolgen, G-CSF ist in Ausnahmefällen notwendig. Die Prognose ist insbesondere bei der isolierten Form gut, die Autoantikörper verschwinden meist in den ersten Lebensjahren. Auch bei der chronisch-benignen idiopathischen Neutropenie sind schwere Infektionen selten. Hier zeigt sich ein protektiver Anstieg der Neutrophilenzahlen bei Infektionen. Eine spezifische Therapie ist meist nicht nötig, die Prognose ist gut.
Die schwere angeborene Neutropenie wird im 1. Lebensjahr symptomatisch mit schweren bakteriellen Infektionen, Pneumonien, Otitiden, Stomatitiden, Abszessen, Haut- und Schleimhautulzerationen. Antibiotikatherapie von Infektionen und –prophylaxe sind notwendig, subkutan verabreichter Granulozyten-Kolonie-stimulierender Faktor (G-CSF) führt zu einer subnormalen Neutrophilenzahl. 5-10% der Kinder zeigen aber kein Ansprechen auf G-CSF. Einzig kurative Therapie ist derzeit die Stammzell- bzw. Knochenmark-Transplantation (SCT/KMT). Unter Antibiotikaprophylaxe und G-CSF ist ein Erreichen des 2. Dezenniums beschrieben, ohne allogene SCT/KMT ist die Prognose aber infaust. Es besteht ein erhöhtes Risiko für die Entstehung einer akuten myeloischen Leukämie (ca. 10%).
Die zyklische Neutropenie zeigt eine variable Symptomatik in Abhängigkeit der Dauer und Schwere der Neutropenie: schwere bakterielle Infektionen, Pneumonien, Abszesse, Lymphadenitis, Stomatitis, Haut- und Schleimhautulzerationen oder nur Fieberschübe können auftreten. Eine entsprechende antibiotische Therapie von Infektionen ist notwendig, durch Gabe von G-CSF lassen sich die Neutrophilenzahlen stabilisieren. Eine antibiotische Prophylaxe ist bei normalen Neutrophilenzahlen nicht erforderlich. Die Prognose ist unter G-CSF gut, die Dauertherapie mit G-CSF erfordert eine Überwachung. Mit einer Ausheilung nach dem zehnten Lebensjahr kann gerechnet werden. Eine maligne Transformation hämatopoetischer Stammzellen wird nicht beobachtet.
Das Shwachman-Diamond-Syndrom zeichnet sich durch eine exokrine Pankreasinsuffizienz (Dystrophie, Durchfälle) mit Infektanfälligkeit bis hin zu septischen Infektionen (Otitis, Pneumonie, Osteomyelitis, Sepsis) aus. Neben der Neutropenie kann eine Thrombozytopenie oder Anämie bestehen. Weitere Symptome sind ekzematös-ichthyosiforme Hauterscheinungen, Kleinwuchs (metaphysäre Chondrodysplasie insbesondere an Femur und Rippen), eine renal tubuläre Funktionsstörung, Hepatopathie und Hepatomegalie. Die Therapie besteht aus der Substitution von Pankreasenzymen und der Gabe von G-CSF. Antibiotika werden bei bakteriellen Infektionen verabreicht, eine antibiotische Prophylaxe ist oft verzichtbar. Bei therapierefraktären Fällen ist eine SCT/KMT zu erwägen. Die Prognose ist durch eine hohe infektassoziierte Mortalität und ein erhöhtes Risiko für maligne Entartung (myeloische Transformation) beeinträchtigt.
Die Glykogenose Typ 1b zeichnet sich durch Hepatomegalie, Nephromegalie, Hypoglykämie, Hyperlipidämie, Hyperurikämie, Kleinwuchs und auch durch eine
Neutropenie aus. Zudem bestehen häufig chronisch entzündliche Darmerkrankungen. Im Glukagontest kommt es zu einem unzureichenden Glukosanstieg. Die Diagnose wird bestätigt durch enzymatische Untersuchung eines Leberbiopsates. Die Neutropenie zeigt ein Ansprechen auf G-CSF. Die Prognose ist durch den Stoffwechseldefekt bestimmt.
1. Ancliff P.J., R.E. Gale, R. Liesner, I.M. Hann, D.C. Linch: Mutations in the ELA2 gene encoding neutrophil elastase are present in most patients with sporadic severe congenital neutropenia but only in some patients with the familial form of the disease. Blood 98, 2645-2650 (2001).
2. Boerkoel C.F., H. Takashima, J. John et al.: Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 30, 215-220 (2002).
3. Cossu F., T.J. Vulliamy, A. Marrone et al.: A novel DKC1 mutation, severe combined immunodeficiency (T+B-NK-SCID) and bone marrow transplantation in an infant with Hoyeraal-Hreidarsson syndrome. Brit J Haematol 119, 765-768 (2002).
4. Dale D.C.: Immune and idiopathic neutropenia. Curr Opin Hematol 5, 33-36 (1998).
5. Dale D.C., R.E. Person, A.A. Bolyard et al.: Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood 96, 2317-2322 (2000).
6. Devriendt K., A.S. Kim,G. Mathijs et al.: Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet 27, 313-317 (2001).
7. Dror Y., H.M. Freedman : Shwachman-Diamond syndrome. Brit J Haematol 118, 701-713 (2002)
8. Hernandez P.A., R.J. Gorlin, J.N. Lukens et al.: Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet 34, 70-74 (2003).
9. Horwitz M., K.F. Bendon, R.E. Person, A.G. Aprikayan, D.C. Dale: Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic hematopoiesis. Nat Genet 23, 433-436 (1999).
10. Person R.E., F.Q. Li, Z. Duan et al.: Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet 34, 308-312 (2003).
11. Welte K., L.A. Boxer: Severe chronic neutropenia: pathophysiology and therapy. Semin Hematol 34, 267-278 (1997).
Tab. 1: Differentialdiagnosen bei Mangel an neutrophilen Granulozyten im Kindes- und Jugendalter.
HÄMATOLOGISCHE URSACHEN UND IMMUNDEFEKTE |
| Morbus Kostmann und andere Formen der schweren angeborenen Neutropenie
Shwachman-Diamond-Syndrom Zyklische Neutropenie Aplastische Anämie Myelodysplastisches Syndrom Retikuläre Dysgenesie Hyper-IgM-Syndrom Schwerer kombinierter Immundefekt (SCID) mit Graft-versus-Host-Disease Familiäre hämophagozytierende Lymphohistiozytose (FHL) Leukämie Lymphome und andere, das Knochenmark infiltrierende Malignomeretikuläre Dysgenesie Wiskott-Aldrich-Syndrom Chediak-Higashi-Syndrom Dyskeratosis congenita / Hoyeraal-Hreidarsson-Syndrom Knorpel-Haar-Hypoplasie Immuno-ossäre Dysplasie Schimke WHIM-Syndrom (Myelokathexis) |
| AUTOIMMUNERKRANKUNGEN |
| Autoimmunneutropenie
Alloimmunneutropenie (konnatal) Systemischer Lupus erythematodes (SLE) Sharp-Syndrom Makrophagenaktivierungssyndrom (MAS) |
| INFEKTIONEN |
| HIV
CMV Parvo-B19 EBV Hepatitis Malaria |
| METABOLISCHE URSACHEN |
| Vitamin B12 Mangel
Folsäuremangel Glykogenose Typ 1b Morbus Gaucher Transcobalaminmangel Propionazidämie Carbamylphosphatsynthase-Defekt Pearson-Syndrom Barth-Syndrom (3-Methyl-Glutaconazidurie Typ II) |
| WEITERE URSACHEN |
| Toxine
Medikamente Chemotherapie Bestrahlung |
Tab. 2: : Immundefekte als Ursache eines chronischen oder rezidivierenden Mangels an neutrophilen Granulozyten im frühen Lebensalter. Abkürzungen: OMIM = Online Mendelian Inheritance in Men; Datenbank des National Institute of Health (NIH) und der Johns Hopkins University in den USA zu genetischen Erkrankungen des Menschen (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM; die angegebenen Nummern entsprechen den Einträgen im McKusick-Verzeichnis).
Krankheit |
Vererbung |
Molekularer Defekt |
Pathogenese | OMIM Eintrag |
| schwere angeborene Neutropenie | autosomal- dominant, sporadisch | Mutationen im Gen der Neutrophilen- Elastase ELA2 oder des Protoonkogens GFI1 | vermehrte Expression bzw. erhöhte Aktivität von ELA2 → gesteigerte Apoptose von (Pro-) Myelozyten bei reduzierter Regenerations- kapazität der Granulopoese im Knochenmark | #202700, 130130, 600871 |
| Kostmann- Syndrom | autosomal-rezessiv | unbekannt | pathogenetisch ungeklärter Ausreifungsstopp der Granulopoese im Stadium der (Pro-) Myelozyten | #202700 |
| X-gebundene schwere angeborene Neutropenie | X-chromosomal rezessiv | konstitutiv- aktivierende Mutation im Wiskott- Aldrich- Syndrom- Protein (WASP) | Störung der Polymersation von Aktin in der Zellmembran → gestörte Zellmotilität, Ausreifungsstopp der Granulopoese auf der Ebene von (Pro-) Myelozyten | #300299, 300392.0012 |
| Zyklische Neutropenie | autosomal- dominant oder -rezessiv | Mutationen im Gen der Neutrophilen-Elastase ELA2 | vermehrte Expression bzw. erhöhte Aktivität von ELA2 → gesteigerte Apoptose von (Pro-) Myelozyten bei erhaltener Regenerations- kapazität der Granulopoese im Knochenmark | #162800, 130130 |
| WHIM- Syndrom | autosomal- dominant oder -rezessiv | Mutationen im Chemokin- Rezeptor- Gen CXCR4 | gestörte Signaltransduktion des mutierten Rezeptors → vermindertes Ansprechen auf SDF-1 → reduzierte Auswanderung reifer Neutrophiler aus dem Knochenmark | #193670, 162643 |
| Shwachman- Diamond- Syndrom | autosomal- rezessiv | Mutationen im SBDS (Shwachman- Bodian- Diamond- Syndrom)-Gen | verstärkte Aktivierung von Apoptosesignalwegen in hämatopoetischen Stammzellen, Ursache unklar (gestörter RNA-Stoffwechsel?) | #260400, 607444 |
| Glykogenose Typ I b | autosomal- rezessiv | Mutationen im Glukose -6- Phosphat- Transporter- Gen mit Mangel an Glukose -6- Phosphat- Translokase | Leber: gestörter Abbau von Glykogen zu Glukose mit pathologischer Glykogenspeicherung; Granulozyten: gestörter mikrosomaler Transport von Glukose -6- Phosphat | #232220, 602671 |